The Art of Life
Nature is remarkably complex and offers a plethora of intricate patterns to those who dare to investigate. These patterns can appear in many forms. Here, we will take a look at how all (sequenced) organisms relate to each other when projecting their high-dimensional genome space down to two dimensions.
[1]:
import os
import gzip
import json
import shutil
import functools
from pathlib import Path
import multiprocessing as mp
from concurrent.futures import Future, as_completed, ProcessPoolExecutor
import numpy as np
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
from matplotlib.patches import Patch
import requests
from requests_futures.sessions import FuturesSession
import khmer
import metagenompy
from Bio import SeqIO
import umap
import umap.plot
import joblib
from tqdm.auto import tqdm
[2]:
%matplotlib inline
umap.plot.output_notebook()
[3]:
CPU_COUNT = 10
DATA_DIR = Path("genome_data")
Retrieve Complete Genomes
Before analyzing the genomes, we need to download them. Here, we are going to download (a subset of) all genomes available on RefSeq.
[4]:
genome_dir = DATA_DIR / "genomes"
genome_dir.mkdir(parents=True, exist_ok=True)
[5]:
# download assembly info
url = (
"https://ftp.ncbi.nlm.nih.gov/genomes/ASSEMBLY_REPORTS/assembly_summary_refseq.txt"
)
path_assembly = DATA_DIR / "assembly_information.tsv"
if not path_assembly.exists():
resp = requests.get(url, stream=True, allow_redirects=True)
resp.raw.read = functools.partial(resp.raw.read, decode_content=True)
with tqdm.wrapattr(resp.raw, "read", desc="Download assembly info") as resp_raw:
with path_assembly.open("wb") as fd:
shutil.copyfileobj(resp_raw, fd)
[6]:
# parse assembly info
df_assembly = pd.read_csv(path_assembly, sep="\t", skiprows=1)
# cleaning
df_assembly.rename(
columns={df_assembly.columns[0]: df_assembly.columns[0].lstrip("#").lstrip()},
inplace=True,
)
# subsetting
df_assembly = df_assembly[
(df_assembly["ftp_path"] != "na")
& (df_assembly["genome_rep"] == "Full")
& df_assembly["excluded_from_refseq"].isna()
& (df_assembly["assembly_level"] == "Complete Genome")
]
# summary
print(df_assembly.shape)
df_assembly.head()
(36277, 23)
[6]:
| assembly_accession | bioproject | biosample | wgs_master | refseq_category | taxid | species_taxid | organism_name | infraspecific_name | isolate | ... | genome_rep | seq_rel_date | asm_name | submitter | gbrs_paired_asm | paired_asm_comp | ftp_path | excluded_from_refseq | relation_to_type_material | asm_not_live_date | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 18 | GCF_000002515.2 | PRJNA12377 | SAMEA3138170 | NaN | representative genome | 28985 | 28985 | Kluyveromyces lactis | strain=NRRL Y-1140 | NaN | ... | Full | 2004/07/02 | ASM251v1 | Genolevures Consortium | GCA_000002515.1 | different | https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/0... | NaN | NaN | na |
| 24 | GCF_000002725.2 | PRJNA15564 | SAMEA3138173 | NaN | representative genome | 347515 | 5664 | Leishmania major strain Friedlin | strain=Friedlin | NaN | ... | Full | 2011/02/14 | ASM272v2 | Friedlin Consortium | GCA_000002725.2 | identical | https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/0... | NaN | NaN | na |
| 25 | GCF_000002765.5 | PRJNA148 | SAMN00102897 | NaN | representative genome | 36329 | 5833 | Plasmodium falciparum 3D7 | NaN | 3D7 | ... | Full | 2016/04/07 | GCA_000002765 | Plasmodium falciparum Genome Sequencing Consor... | GCA_000002765.3 | different | https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/0... | NaN | NaN | na |
| 34 | GCF_000002985.6 | PRJNA158 | SAMEA3138177 | NaN | reference genome | 6239 | 6239 | Caenorhabditis elegans | strain=Bristol N2 | NaN | ... | Full | 2013/02/07 | WBcel235 | C. elegans Sequencing Consortium | GCA_000002985.3 | different | https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/0... | NaN | NaN | na |
| 65 | GCF_000005825.2 | PRJNA224116 | SAMN02603086 | NaN | na | 398511 | 79885 | Alkalihalobacillus pseudofirmus OF4 | strain=OF4 | NaN | ... | Full | 2010/12/15 | ASM582v2 | Center for Genomic Sciences, Allegheny-Singer ... | GCA_000005825.2 | identical | https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/0... | NaN | NaN | na |
5 rows × 23 columns
[7]:
def download_genome(row, target_dir, session):
name = row.ftp_path.rsplit("/", 1)[-1]
fname = f"{name}_genomic.fna.gz"
path = target_dir / fname
meta_path = f"{path}.json"
url = f"{row.ftp_path}/{fname}"
if path.exists():
# print("Using cache for", url)
future = Future()
future.set_result("foo")
else:
# print("Downloading", url)
future = session.get(url)
future.path = path
future.meta_path = meta_path
future.accession = row.assembly_accession
future.taxid = row.taxid
return future
To speed up the download (which is IO bound), we will distribute this task over multiple threads.
[8]:
with FuturesSession(max_workers=CPU_COUNT) as session:
futures = df_assembly.apply(
download_genome, axis=1, args=(genome_dir, session)
).tolist()
for future in tqdm(
as_completed(futures), total=len(futures), desc="Download genomes"
):
resp = future.result()
if isinstance(resp, requests.models.Response):
with open(future.meta_path, "w") as fd:
json.dump({"accession": future.accession, "taxid": future.taxid}, fd)
with open(future.path, mode="wb") as fd:
fd.write(resp.content)
Compute Genomic Features
We are going to represent DNA sequences by their kmer count profile. In addition, we will compute further statistics, such as, for example, the number of base pairs in each genome.
Helper functions
[9]:
def load_files(path):
"""Retrieve sequence and metadata for given entry."""
with gzip.open(path, "rt") as fd:
record_list = list(SeqIO.parse(fd, "fasta"))
# aggregate sequences
seq = ""
for record in record_list:
seq += str(record.seq)
seq = seq.upper()
# get metadata
path_meta = f"{path}.json"
with open(path_meta) as fd:
metadata = json.load(fd)
return seq, metadata
[10]:
def compute_kmer_counts(seq, k=3):
"""
https://github.com/dib-lab/khmer/blob/master/examples/python-api/exact-counting.py
"""
# setup counter
nkmers = 4**k
tablesize = nkmers + 10
cg = khmer.Countgraph(k, tablesize, 1)
cg.set_use_bigcount(True) # increase max count from 255 to 65535
# count kmers
cg.consume(seq)
# return formatted output
return {cg.reverse_hash(i): cg.get(i) for i in range(nkmers)}
[11]:
def parse_entry(path):
"""Do all computations for single genome file."""
assert str(path).endswith("_genomic.fna.gz"), path
# parse entry
seq, meta = load_files(path)
# handle meta information
meta["genome_size"] = len(seq)
# count kerms
kmer_counts = compute_kmer_counts(seq, k=5)
return meta, kmer_counts
Parse data
As computing these features is CPU bound, we are going to make use of multiprocessing.
[12]:
kmer_data = {}
metadata = []
with ProcessPoolExecutor(
max_workers=CPU_COUNT, mp_context=mp.get_context("fork")
) as executor:
futures = [
executor.submit(parse_entry, path)
for path in genome_dir.iterdir()
if str(path).endswith("_genomic.fna.gz")
]
for future in tqdm(as_completed(futures), total=len(futures), desc="Parse genomes"):
# compute stuff
meta, kmer_counts = future.result()
# keep results
metadata.append(meta)
id_ = meta["accession"]
assert id_ not in kmer_data
kmer_data[id_] = kmer_counts
Determine taxonomic lineage for each entry
To investigate how different types of organisms relate to each other, we will characterize each organisms by its taxonomic rank.
[13]:
# list of which ranks to consider
rank_list = ["species", "phylum", "clade", "kingdom", "superkingdom"]
[14]:
graph = metagenompy.generate_taxonomy_network(auto_download=True)
Parsing names: 100%|██████████| 3532357/3532357 [00:04<00:00, 721381.84it/s]
Parsing nodes: 100%|██████████| 2388279/2388279 [00:21<00:00, 112286.89it/s]
[15]:
df_meta = pd.DataFrame(metadata).set_index("accession")
df_meta["taxid"] = df_meta["taxid"].astype(str)
df_meta = metagenompy.classify_dataframe(graph, df_meta, rank_list=rank_list)
Classifying: 100%|██████████| 5/5 [00:02<00:00, 2.22it/s]
Save results
[16]:
df_meta.to_csv(DATA_DIR / "metadata.csv.gz")
df_meta.head()
[16]:
| taxid | genome_size | species | phylum | clade | kingdom | superkingdom | |
|---|---|---|---|---|---|---|---|
| accession | |||||||
| GCF_002191655.1 | 29459 | 3312719 | Brucella melitensis | Proteobacteria | <NA> | <NA> | Bacteria |
| GCF_000852745.1 | 103881 | 17266 | Potato yellow vein virus | Kitrinoviricota | Riboviria | Orthornavirae | Viruses |
| GCF_002197575.1 | 1983777 | 14964 | Avian metaavulavirus 15 | Negarnaviricota | Riboviria | Orthornavirae | Viruses |
| GCF_006384535.1 | 2588128 | 59514 | Gordonia phage Barb | Uroviricota | Duplodnaviria | Heunggongvirae | Viruses |
| GCF_000025865.1 | 547558 | 2012424 | Methanohalophilus mahii | Euryarchaeota | Stenosarchaea group | <NA> | Archaea |
[17]:
df_kmer = pd.DataFrame(kmer_data)
df_kmer.index.name = "kmer"
df_kmer.to_csv(DATA_DIR / "kmer_counts.csv.gz")
df_kmer.head()
[17]:
| GCF_002191655.1 | GCF_000852745.1 | GCF_002197575.1 | GCF_006384535.1 | GCF_000025865.1 | GCF_000019085.1 | GCF_000800395.1 | GCF_000861705.1 | GCF_000879055.1 | GCF_014127105.1 | ... | GCF_002448155.1 | GCF_016026895.1 | GCF_003595175.1 | GCF_019192625.1 | GCF_018289355.1 | GCF_007954485.1 | GCF_016403105.1 | GCF_011765625.1 | GCF_900638255.1 | GCF_015571675.1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| kmer | |||||||||||||||||||||
| AAAAA | 7574 | 120 | 51 | 3 | 15189 | 37345 | 2727 | 63 | 13 | 6061 | ... | 1467 | 16316 | 4522 | 22659 | 36468 | 42322 | 12171 | 33204 | 7380 | 23280 |
| AAAAT | 7646 | 160 | 63 | 6 | 11130 | 27333 | 1677 | 44 | 8 | 6781 | ... | 1625 | 11873 | 2734 | 17629 | 28855 | 34908 | 11573 | 28167 | 6670 | 18076 |
| AAAAC | 7884 | 72 | 33 | 11 | 7672 | 12339 | 2710 | 47 | 11 | 7593 | ... | 3149 | 13131 | 4546 | 16801 | 19309 | 22568 | 10829 | 20225 | 8343 | 17542 |
| AAAAG | 9115 | 77 | 29 | 4 | 10460 | 24769 | 3071 | 40 | 7 | 7159 | ... | 2251 | 6911 | 3583 | 12260 | 26820 | 23141 | 8646 | 18385 | 4012 | 12757 |
| AAATA | 4774 | 119 | 56 | 11 | 9577 | 21599 | 1022 | 19 | 12 | 3594 | ... | 1328 | 6824 | 2166 | 11850 | 28194 | 27460 | 5007 | 23131 | 4916 | 11996 |
5 rows × 36277 columns
Data overview
Before analyzing the data in more depth, we check whether reasonable kmer counts have been generated and whether the taxonomic classification worked out.
[18]:
# did the kmer counting work
max_count_fraction = (df_kmer >= 65535).sum().sum() / (
df_kmer.shape[0] * df_kmer.shape[1]
)
print(f"{max_count_fraction * 100:.2f}% of kmer counts have reached numeric maximum")
0.08% of kmer counts have reached numeric maximum
[19]:
# how many NAs are in our taxonomic metadata
df_meta.isna().sum()
[19]:
taxid 0
genome_size 0
species 27
phylum 2041
clade 15606
kingdom 26965
superkingdom 27
dtype: int64
Additionally, we can briefly look at some interesting summary statistics.
[20]:
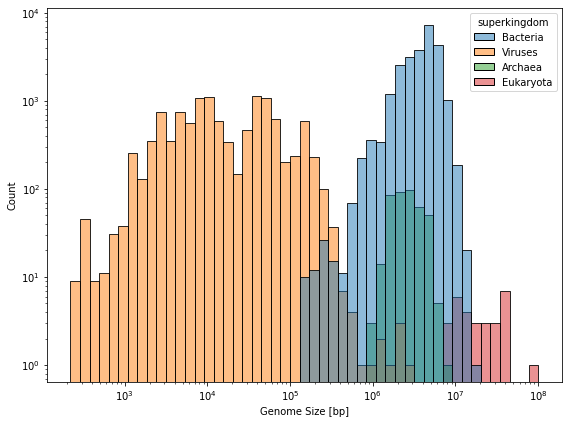
fig, ax = plt.subplots(figsize=(8, 6))
sns.histplot(data=df_meta, x="genome_size", hue="superkingdom", log_scale=True, ax=ax)
ax.set_xlabel("Genome Size [bp]")
ax.set_yscale("log")
fig.tight_layout()
fig.savefig(DATA_DIR / "genome_size_hist.pdf")
Kmer statistics
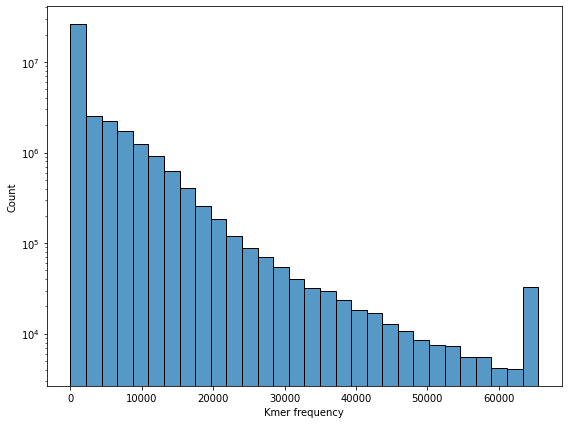
Before reducing the dimensionality of the kmer space, let’s look at a few of its features.
Let’s start by checking the overall kmer count distribution. We can observe a peak at \(0\) as well as a peak at the numeric kmer count maximum of \(65535\).
[21]:
fig, ax = plt.subplots(figsize=(8, 6))
sns.histplot(data=df_kmer.values.ravel(), bins=30, ax=ax)
ax.set_xlabel("Kmer frequency")
ax.set_yscale("log")
fig.tight_layout()
fig.savefig(DATA_DIR / "kmer_count_hist.pdf")
We then continue by looking at the most/least common kmers averaged over all organisms.
[22]:
kmer_counts = df_kmer.median(axis=1)
kmer_counts = kmer_counts[kmer_counts > 0]
print("Most common kmers:")
print(kmer_counts.head())
print()
print("Least common (non-zero) kmers:")
print(kmer_counts.tail())
Most common kmers:
kmer
AAAAA 8464.0
AAAAT 6449.0
AAAAC 6518.0
AAAAG 6762.0
AAATA 4391.0
dtype: float64
Least common (non-zero) kmers:
kmer
GCCCC 2177.0
GCCGC 3081.0
GCGCC 2676.0
GGACC 1526.0
GGCCC 1114.0
dtype: float64
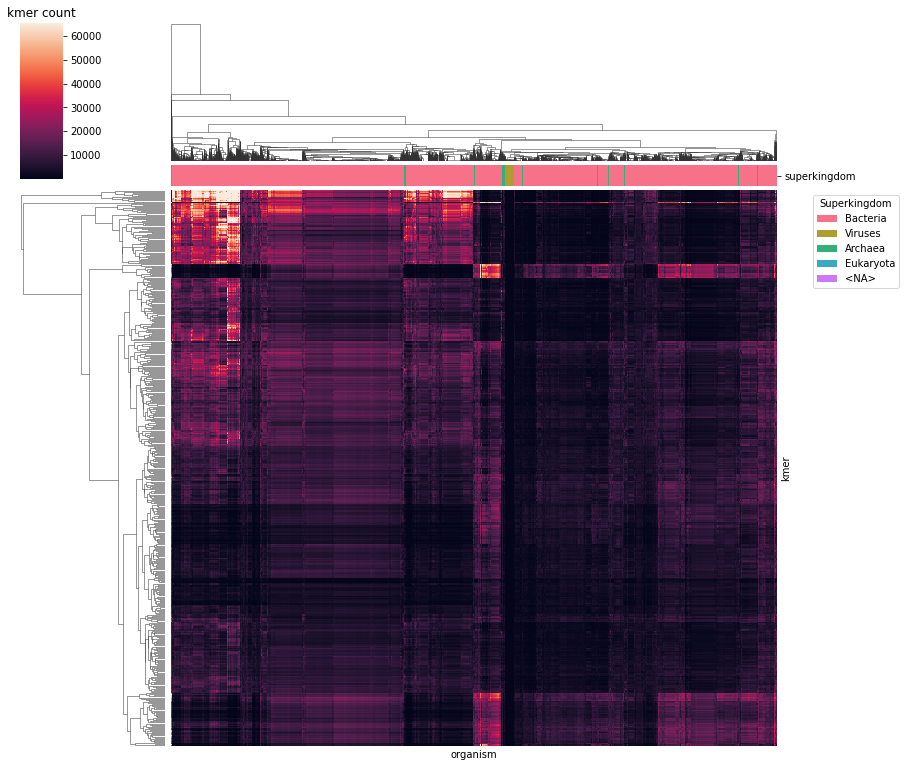
Finally, we can enjoy a clustered heatmap.
[23]:
%%time
# retain rows with non-zero entries and columns with not-low entries
df_kmer_sub = df_kmer.loc[(df_kmer > 0).any(axis=1), (df_kmer.median(axis=0) > 10)]
df_kmer_sub.columns.rename("organism", inplace=True)
# generate column color map
rank_colors = {
rank: sns.color_palette("husl", df_meta["superkingdom"].nunique(dropna=False))[i]
for i, rank in enumerate(df_meta["superkingdom"].unique())
}
rank_cmap = df_meta.loc[df_kmer_sub.columns, "superkingdom"].map(rank_colors)
# create plot
g = sns.clustermap(
df_kmer_sub,
col_colors=rank_cmap,
rasterized=True,
figsize=(12, 12),
)
g.cax.set_title("kmer count")
g.ax_heatmap.tick_params(bottom=False, labelbottom=False, right=False, labelright=False)
g.ax_heatmap.legend(
handles=[Patch(facecolor=color, label=name) for name, color in rank_colors.items()],
title="Superkingdom",
bbox_to_anchor=(1.05, 1),
loc="upper left",
)
fig.savefig(DATA_DIR / "kmer_heatmap.pdf", dpi=300)
/cluster/work/bewi/nss/apps/gcc-6.3.0/conda/4.8.3/lib/python3.8/site-packages/seaborn/matrix.py:654: UserWarning: Clustering large matrix with scipy. Installing `fastcluster` may give better performance.
warnings.warn(msg)
CPU times: user 2min 40s, sys: 6.02 s, total: 2min 46s
Wall time: 2min 47s
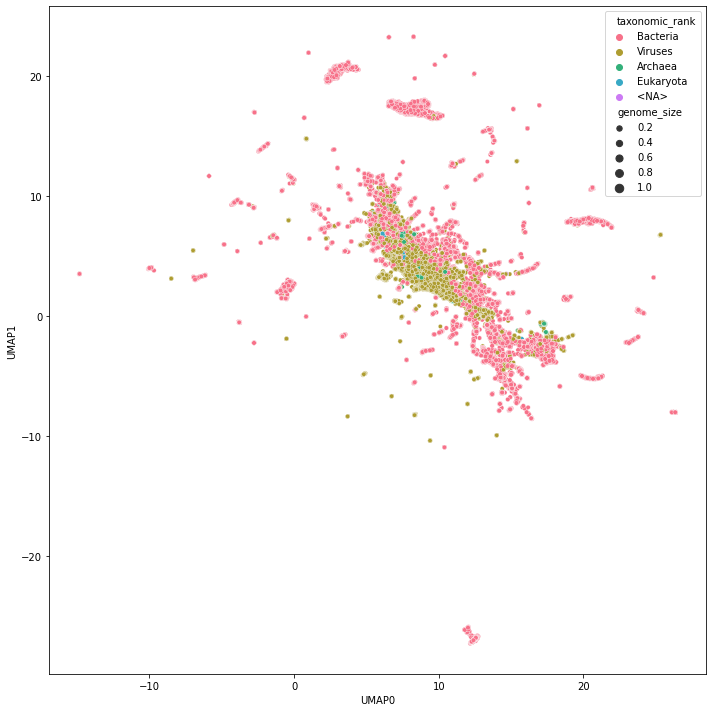
Visualize Projected Genome Space
Finally, we can project the high-dimensional kmer space to two dimensions and explore its topology.
[24]:
reducer = umap.UMAP(
metric="cosine",
random_state=42,
low_memory=True,
verbose=True,
n_neighbors=100,
n_jobs=min(CPU_COUNT, 8),
)
[25]:
reducer.fit(df_kmer.T)
UMAP(angular_rp_forest=True, metric='cosine', n_jobs=8, n_neighbors=100, random_state=42, verbose=True)
Wed Jan 5 23:36:01 2022 Construct fuzzy simplicial set
Wed Jan 5 23:36:01 2022 Finding Nearest Neighbors
Wed Jan 5 23:36:01 2022 Building RP forest with 15 trees
Wed Jan 5 23:36:06 2022 NN descent for 15 iterations
1 / 15
2 / 15
3 / 15
Stopping threshold met -- exiting after 3 iterations
Wed Jan 5 23:37:06 2022 Finished Nearest Neighbor Search
Wed Jan 5 23:37:11 2022 Construct embedding
Wed Jan 5 23:38:22 2022 Finished embedding
[25]:
UMAP(angular_rp_forest=True, metric='cosine', n_jobs=8, n_neighbors=100, random_state=42, verbose=True)
[26]:
joblib.dump(reducer, DATA_DIR / "umap_model.joblib")
Wed Jan 5 23:38:27 2022 Worst tree score: 0.96907131
Wed Jan 5 23:38:27 2022 Mean tree score: 0.97277063
Wed Jan 5 23:38:27 2022 Best tree score: 0.97499793
Wed Jan 5 23:38:37 2022 Forward diversification reduced edges from 3627700 to 258377
Wed Jan 5 23:38:40 2022 Reverse diversification reduced edges from 258377 to 257088
/cluster/home/kimja/.local/lib/python3.8/site-packages/scipy/sparse/_index.py:125: SparseEfficiencyWarning: Changing the sparsity structure of a csr_matrix is expensive. lil_matrix is more efficient.
self._set_arrayXarray(i, j, x)
Wed Jan 5 23:38:42 2022 Degree pruning reduced edges from 297740 to 297717
Wed Jan 5 23:38:42 2022 Resorting data and graph based on tree order
Wed Jan 5 23:38:42 2022 Building and compiling search function
[26]:
['genome_data/umap_model.joblib']
Static visualization
We can look at a static image…
[27]:
def format_taxonomy_string(taxonomy, rank_list=["superkingdom", "kingdom"]):
"""Convert classification columns to readable string."""
return ";".join(str(taxonomy[rank]) for rank in rank_list)
[28]:
embedding = reducer.transform(df_kmer.T)
df_umap = pd.DataFrame(embedding)
df_umap.columns = ("UMAP0", "UMAP1")
df_umap["accession"] = df_kmer.columns
df_umap["taxonomic_rank"] = df_umap["accession"].apply(
lambda x: format_taxonomy_string(df_meta.loc[x], rank_list=["superkingdom"])
)
df_umap.set_index("accession", inplace=True)
df_umap["genome_size"] = df_meta["genome_size"]
df_umap.to_csv(DATA_DIR / "umap.csv.gz")
print(df_umap.shape)
df_umap.head()
(36277, 4)
[28]:
| UMAP0 | UMAP1 | taxonomic_rank | genome_size | |
|---|---|---|---|---|
| accession | ||||
| GCF_002191655.1 | 18.933571 | 1.356702 | Bacteria | 3312719 |
| GCF_000852745.1 | 7.202410 | 5.276213 | Viruses | 17266 |
| GCF_002197575.1 | 7.665549 | 3.549558 | Viruses | 14964 |
| GCF_006384535.1 | 17.502321 | -1.682356 | Viruses | 59514 |
| GCF_000025865.1 | 8.906975 | 5.523824 | Archaea | 2012424 |
[29]:
fig, ax = plt.subplots(figsize=(10, 10))
sns.scatterplot(
data=df_umap,
x="UMAP0",
y="UMAP1",
hue="taxonomic_rank",
size="genome_size",
rasterized=True,
palette=sns.color_palette("husl", df_umap["taxonomic_rank"].nunique()),
ax=ax,
)
fig.tight_layout()
fig.savefig(DATA_DIR / "umap.pdf", dpi=300)
Interactive visualization
…but also pan and zoom around in an interactive view.
[30]:
hover_data = df_meta.loc[df_kmer.columns].rename_axis("accession").reset_index()
hover_data["genome_size"] = hover_data["genome_size"].apply(lambda x: f"{x:,} bp")
[31]:
p = umap.plot.interactive(
reducer,
labels=df_umap["taxonomic_rank"].reset_index(drop=True),
theme="fire",
hover_data=hover_data,
point_size=2,
)
umap.plot.show(p)